Artículo de investigación Centro Dermatológico Dr. Úraga | Vol 6 | Nº 1 2024
Neurofibromatosis Segmentaria
Camila Alejandra Félix Caviedes,* Yoselin Aracely Chamorro Gaón,*
Oswaldo Patricio Freire Murgueytio**
Oswaldo Patricio Freire Murgueytio**
Cuadro Clínico
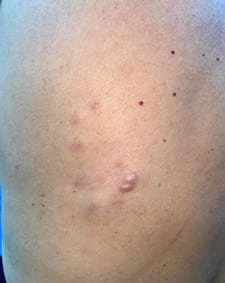
Se presenta el caso de una paciente femenina de 70 años, sin antecedentes patológicos personales ni familiares de importancia, que acude a consulta de dermatología con un cuadro cutáneo de 8 años de evolución caracterizado por un nódulo exofítico, blando, eucrómico, liso, no móvil, en región subescapular izquierda, que le ocasiona ardor y prurito. Además, se acompaña a su alrededor de otros nódulos subcutáneos, blandos, pequeños, de color violáceo asintomáticos (Foto 1). No presentó otras alteraciones al examen físico.
Nódulo exofítico eucrómico rodeado por nódulos subcutáneos violáceos en región escapular izquierda
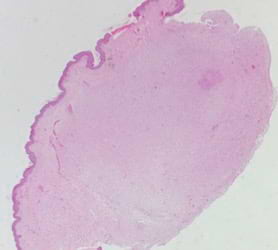
Se realizó una biopsia incisional de la lesión de mayor tamaño que reportó ortoqueratosis y epidermis irregular. A nivel dérmico: lesión nodular, bien delimitada, constituida por proliferación de células alargadas que conservan la relación núcleo-citoplasma, acompañada de algunos mastocitos y vasos congestivos, sin signos de malignidad (Foto 2). Con base en estos hallazgos histológicos se catalogó como neurofibroma. En la exploración oftalmológica, no se evidenciaron nódulos de Lisch. La valoración neurológica no presentó alteraciones. También se realizó una tomografía computarizada (TC) craneal, una ecografía abdominal y una radiografía de tórax sin encontrar hallazgos significativos. No existen familiares afectados con lesiones similares. Debido a esto y a que los neurofibromas se encontraban localizados en un segmento del cuerpo sin otros signos de enfermedad, se diagnosticó de neurofibromatosis segmentaria. Se realizó la exéresis completa de la lesión exofítica con vigilancia periódica de las otras lesiones.
DiscusiónLa neurofibromatosis es una genodermatosis que afecta la piel, el sistema nervioso o ambos y fue descrita en 1882 por Von Recklinghausen. 1 Se han realizado varias clasificaciones, una de ellas es la de Ricardi que la clasifica en 7 tipos, entre las que se encuentra la neurofibromatosis segmentaria, también conocida como neurofibromatosis tipo V o neurofibromatosis localizada en mosaico, que es una forma rara de esta enfermedad en la que manchas cafe-au-lait y/o neurofibromas aparecen limitadas a una zona del cuerpo, sin trasfondo familiar.1,2 Tiene una prevalencia baja de 0,0014 a 0,002% y ocurre dos veces más en mujeres y hay un pico bimodal de aparición entre los 10 y 30 años y entre los 50 y 70 años.
El mecanismo de transmisión de esta enfermedad no está claro, pero se lo atribuye a una mutación somática poscigótica temprana de las células primitivas de la cresta neural, que afecta al gen NF1, situado en la región pericentromérica del brazo largo del cromosoma 17.
Los neurofibromas son la manifestación cutánea más común, observándose en el 70% de los pacientes, seguido de las manchas cafe-au-lait en el 44% de los casos y las efélides axilares o inguinales en el 20%, siempre asociadas a manchas cafe-au-lait.1 Según las manifestaciones clínicas, los pacientes se pueden dividir en cuatro grupos: con lesiones solo pigmentarias, con solo neurofibromas, con lesiones pigmentarias y neurofibromas y con neurofibromas plexiformes aisladas. 3 En la mayoría de los casos, las lesiones siguen las líneas de Blaschko y suelen ser unilaterales y ocupan un solo dermatoma, pero en el 6% de los casos son bilaterales.
La afectación sistémica es rara, a excepción de pacientes con neurofibromas plexiformes. Pueden encontrarse neurofibromas en el abdomen, el mediastino y el tracto urogenital. Las neoplasias malignas asociadas incluyen cáncer de mama, cáncer de colon, cáncer gástrico, cáncer de pulmón, linfoma de Hodgkin, tumor de la vaina del nervio periférico, melanoma maligno, siendo los dos últimos los más comunes. Debido a esta frecuencia de asociaciones, es importante realizar un estudio sistémico en estos pacientes, que incluya anamnesis, exploración física, evaluación oftalmológica, exploración neurológica (TC craneal y resonancia magnética) y estudio de extensión (radiografía de tórax, ecografía abdominal y mapa óseo), así como un seguimiento multidisciplinar del paciente a largo plazo.
Para el diagnóstico se debe correlacionar clínicamente con los hallazgos histopatológicos de los neurofibromas.4 El diagnóstico diferencial de los neurofibromas se debe realizar principalmente con schwannoma, leiomioma cutáneo, dermatofibroma, nevo lipomatoso y anetodermia.
Bibliografía1. Martínez S, Vera Á, Sanz A, Crespo V. Neurofibromatosis segmentaria verdadera. Actas Dermosifiliogr. 2004;95(3), 175-177.
2. Suárez Fernández R, Trasobares L, Medina S, García Rodríguez M. Neurofibromatosis. Med Integr. 2001; 38(2):64–68.
3. Sobjanek, M., Dobosz-Kawałko, M., Michajłowski, I., Pęksa, R., & Nowicki, R. Segmental neurofibromatosis. Postepy dermatologii i alergologii. 2014;31(6), 410–412.
4. Ramos-Espinoza AB, Garza-Tovar TF, Mesa-Garza IG, González-Cabello D, González-Murillo E, Navarrete-Solís y. J. Neurofibromatosis segmentaria verdadera: presentación de un caso. DermatologíaCMQ. 2021;19(2):153-156.