Cuadro Clínico
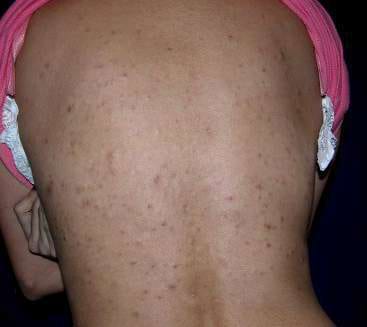
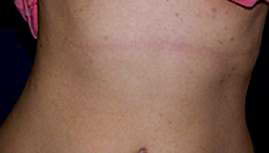
Paciente de sexo femenino, de 27 años de edad sin antecedentes personales de importancia, de nacionalidad ecuatoriana y residente en Guayaquil, consulta por primera vez en octubre del año 2008 por presentar desde su infancia, numerosas lesiones nodulares amarillentas, irregulares, de diverso tamaño que fluctuaban entre 0.2 a 0.5 cm de diámetro, localizadas en espalda (Fig. 1), brazos (Fig. 2), abdomen (Fig. 3) y muslos (Fig. 4). Estas lesiones presentaban superficie lisa, sin tendencia a confluir ni síntomas asociados.
Durante el interrogatorio, la paciente manifestó que su madre presentó el mismo tipo de lesiones ubicadas en los brazos las cuales desaparecieron espontáneamente luego de la adolescencia.
Haga su diagnóstico
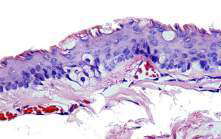
Se solicitó biopsia con estudio histopatológico, el cual reportó: ¨Piel con estructura quística profunda, revestida por epitelio escamoso estratificado cuyo borde libre es festoneado. Presencia de sebocitos en la pared del quiste¨ (Fig. 5).
Discusión
El término Esteatocitoma viene de raíces griegas que significan ¨bolsa de grasa¨. Jaimeson en el año de 1873 fue el primero en reportar un paciente con numerosos quistes repartidos en todo el cuerpo. Posteriormente, Dubreuilh, Auche y Bosellini describieron casos similares. El primero en describir la enfermedad con el nombre de EM fue Pringle en el año 1899.2-4 Hans Gunther en 1917 da vida al término sebocistomatosis al reportar el caso de un joven con múltiples quistes cutáneos..
Su patogénesis sigue siendo incierta pero se la denomina predominantemente como una malformación hamartomatosa del conducto pilosebáceo,5 Plewig, Wolf y Braun Falco lo catalogan como un tumor nevoide de los conductos y de las glándulas sebáceas, que se origina en los folículos sebáceos.6 Su presentación puede ser esporádica o puede ser familiar habiendo sido bien documentada hasta en cinco generaciones lo cual sugiere un patrón de herencia autosómico dominante. 7 Diversos autores como Yoneda et al.8 piensan que el EM puede ser causado por una mutación en el gen de la queratina 17 (K17). Como resultado de esta mutación genética, la red de filamentos intermedios de queratina queda interrumpida.
Las lesiones se presentan como pápulas o nódulos numerosos y recurrentes con ubicación preferencial en zonas con unidades pilosebáceas densamente concentradas como son las áreas antes citadas y con menor frecuencia en genitales o mamas, en cara, dando incluso apariencia de facie leonina9 y cuero cabelludo. La región esternal es la más afectada en varones. Noexiste predilección de sexo ni raza. De acuerdo con la distribución de las lesiones se las ha clasificado como localizadas, generalizadas, faciales, acrales, como elcaso reportado por Jain en un solo pie13 y una variante denominada supurativa.14 Igualmente, Almeida et al.15 y Park et al.,16 reportaron una forma unilateral lineal siendo el caso de Park de carácter congénito y con ubicación nasal. Las lesiones solitarias son esporádicas y se las ha denominado como esteatocistoma simple (ES). El ES oral es un tumor muy raro. Kaya et al. reportaron el primer caso de ES de paladar blando.
Las lesiones tempranas de EM se presentan con apariencia cupuliforme y aspecto translúcido que con el paso de los años toman una coloración amarillenta y, en raras ocasiones pueden presentar un color azul oscuro.18 Son de consistencia elástica o firme así como quística, se ha reportado igualmente su calcificación. Cuando se produce una ruptura espontánea de los quistes va a dar origen a la forma supurativa del EM con inflamación y luego cicatrización residual que se asemeja al acné conglobata.21 La presencia de comedones sin punta visible constituye una asociación característica.11 Las lesiones generalmente son asintomáticas pero pueden ser pruriginosas o dolorosas cuando se infectan.
EM ha sido tratado con éxito con Erbium: YAG láser o láser de CO2, radiofrecuencia y procedimientos quirúrgicos excisionales u otros mínimamente invasivos como aspiración con aguja. La crioterapia ha sido usada pero la posibilidad de cicatrices y discromías residuales limitan su uso. La isotretinoína ha sido preconizada como terapia oral teniendo como ventajas la prevención en la formación de nuevas lesiones y el hecho de que las lesiones pueden reducirse tras la interrupción de la isotretinoína y como desventaja la posibilidad de recurrencia y que los buenos resultados estén limitados a lesiones inflamadas. La desaparición espontánea ha sido reportada pero con remotas posibilidades.
Bibliografía
1. Covello SP, Smith FJ, SillevisSmitt JH, Paller AS, Munro CS, Jonkman MF, et al. Keratin 17 mutations cause either steatocystoma multiplex or pachyonychia congenita type 2. Br JDermatol. 1998;139(3):475-80.
2. Marrugo-Lara J, Hernández-Arana MS, Hernández MM. Esteatocistoma múltiple y quistes eruptivos vellosos. Dermatol Rev Mex. 2018;62:130-136.
3. Georgakopoulos JR, Ighani A, Yeung J. Numerous asyntomatic dermal cyst. Canadian Family Phycician / Le Medicin de famile canadien. 2018;64:892-899.
4. Hemlata T. Karma et al., Steatocystoma Multiplex- A Rare Genetic Disorder. Journal of Clinical and Diagnostic Research. 2013:7: 166-168.
5. Young Shin N, Kang JE, Kim JE, Symkhampa K, Hoe Huh K, Yi WJ, Heo MS,, Lee SS, Choi SC. Steatocystoma multiplex: A case report of a rare entity. Imaging Science in Dentistry 2019; 49: 317-21.
6. Plewig G, Wolff HH, Braun-Falco O. Steatocystoma Multiplex: Anatomic Reevaluation, Electron Microscopy, and Autoradiography Arch Dermatol Res 1982;272:363- 380.