Cuadro Clínico
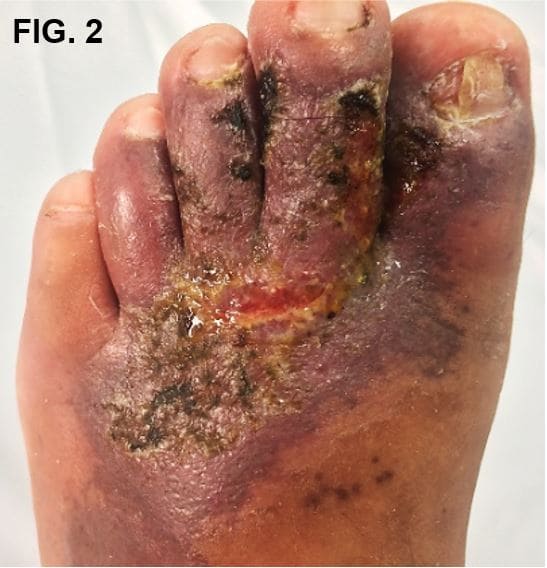
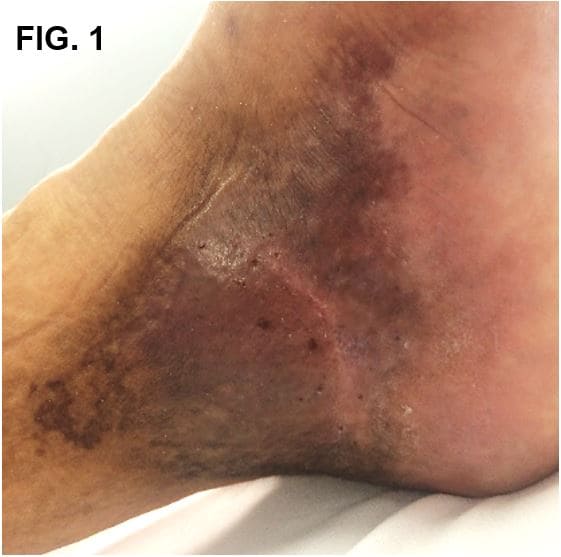
Se presenta el caso de paciente masculino de 33 años con antecedente de trombosis venosa profunda en pierna izquierda hace 9 años complicada con tromboembolia pulmonar. Remitido de cirugía vascular a dermatología por cuadro de 2 años de evolución de mácula hiperpigmentada color ocre en el maléolo interno del pie izquierdo que se extiende a dorso de pie como placa violácea (Fig. 1); que, en 2 meses evoluciona a úlcera de bordes irregulares localizada del primer al cuarto dedo del pie, cubierta de costra melicérica, con presencia de exudado sero-purulento, acompañada de edema e imposibilidad funcional (Fig. 2). Pulsos pedios presentes.
Haga su diagnóstico
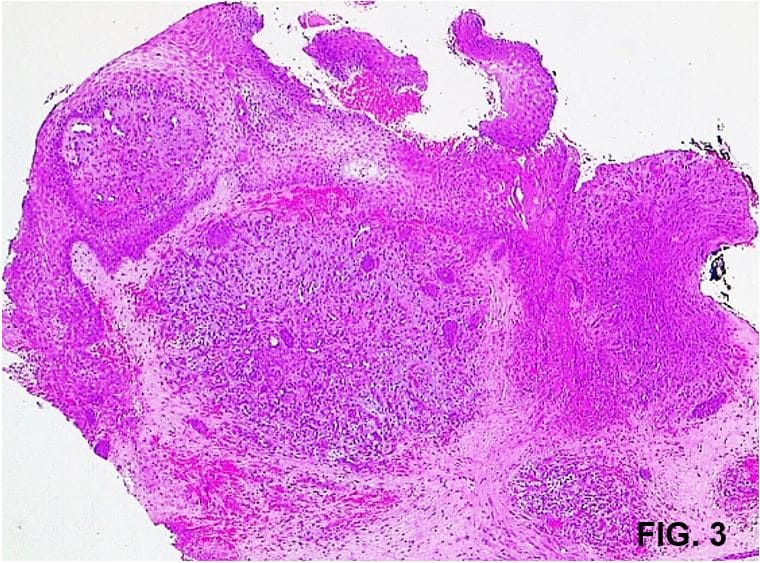
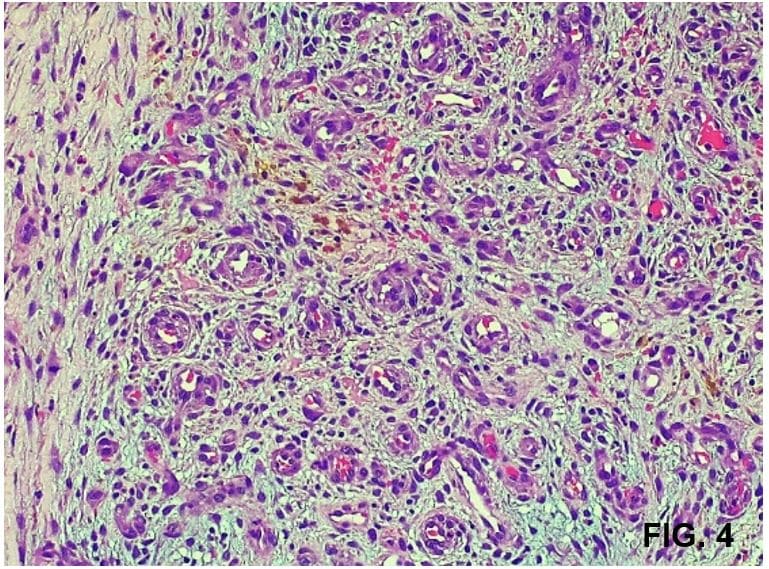
Dentro de los exámenes complementarios, el cultivo de secreción evidencia Enterococcus faecalis sensible a vancomicina; el histopatológico de la lesión reporta proliferación de vasos de pequeño calibre dilatados con endotelio prominente sobre dermis edematosa rodeada de células endoteliales con áreas donde los vasos forman nódulos, eritrocitos extravasados y depósitos de hemosiderina (Fig. 3 y 4). Se consulta nuevamente a cirugía vascular donde realizan angiotomografía con evidencia de compresión de la vena ilíaca izquierda estando ésta entre la arteria ilíaca izquierda y el cuerpo vertebral de L5, llegando al diagnóstico de Síndrome de May-Thurner (SMT) (Fig. 5).
<
Discusión
La acroangiodermatitis es un trastorno vascular cutáneo benigno e infrecuente englobado dentro de las enfermedades angioproliferativas reactivas. La variante tipo Mali ocurre con mayor frecuencia en hombres adultos, distalmente en extremidades inferiores, asociada a insuficiencia venosa crónica (IVC). Debido a que la IVC es la enfermedad vascular más frecuente, es más probable encontrar la AAD como una rara complicación en pacientes con estasis venosa crónica e hipertensión venosa de los miembros inferiores como en este paciente, cuyo desencadenante resultó ser el síndrome de May-Thurner.
El SMT, síndrome de Cockett o síndrome de compresión de la vena iliaca, se refiere más frecuentemente al fenómeno de compresión de la vena ilíaca izquierda por la arteria ilíaca común derecha por encima y la quinta vértebra lumbar por debajo; siendo, el lado izquierdo el más afectado. Ocurre predominantemente en mujeres entre la tercera y quinta década de vida .
La prevalencia es relativamente alta, sin embargo es subdiagnosticada porque en la mayoría de casos es asintomática; y, en un 2 a 5% produce cuadros de IVC (venas varicosas, pigmentación en piernas, úlceras venosas) o trombosis venosa profunda (TVP), debido al daño crónico del tejido venoso producido por la pulsación arterial constante. El tratamiento óptimo incluye eliminar el trombo (trombólisis con catéter o aspiración mecánica) si existe TVP, seguido de la corrección de la estenosis iliaca subyacente mediante dilatación con balón, filtro de vena cava o colocación de stent.
A pesar que la literatura sugiere que el SMT debe considerarse en casos de TVP del miembro inferior izquierdo, en nuestro paciente, que tuvo este antecedente complicado de tromboembolia pulmonar a una edad joven, inicialmente, no se pensó como diagnóstico posible y no recibió el tratamiento correspondiente. Tras la evaluación del trastorno dermatológico cuyo resultado histopatológico reveló AAD tipo Mali, se solicita una nueva valoración por cirugía vascular para indagar la causa subyacente de la IVC, lo que permitió finalmente, descubrir la compresión iliaca, diagnosticar y tratar de manera óptima el SMT, evitando otras complicaciones tromboembólicas y favoreciendo la resolución de la patología cutánea.
La AAD es una dermopatía angioproliferativa rara cuya variante tipo Mali suele iniciar en la extremidad inferior izquierda para posteriormente ser bilateral en respuesta a insuficiencia venosa crónica. En nuestro paciente se identificaron como factores de IVC el SMT y la TVP. En el presente caso y en la mayoría de los publicados, el diagnóstico de la AAD es inicialmente erróneo o se retrasa, ya sea porque sus síntomas son inespecíficos, imitando otras afecciones cutáneas o porque se desconoce su existencia, convirtiéndose en un reto, especialmente si no se considera como posible desencadenante al SMT. Por lo tanto, es necesario un alto grado de sospecha y debe considerarse como diagnóstico diferencial a la ADD ante lesiones angioproliferativas resultantes de insuficiencia venosa y dermatitis de estasis, a fin de evitar tratamientos inadecuados y complicaciones, proceder a la corrección de la causa de base y proporcionar un mejor pronóstico y calidad de vida.
Bibliografía
1. Chea EP, Rutt VL, Levin J, McClain R, Purcell SM. Acroangiodermatitis of Mali and stewart-bluefarb syndrome. Cutis. 2019;103
2. Yosipovitch G, Nedorost ST, Silverberg JI, Friedman AJ, Canosa JM, Cha A. Stasis Dermatitis: An Overview of Its Clinical Presentation, Pathogenesis, and Management. Am J Clin Dermatol. 2023;24.
3. Yang C, Li D, Li Y, Li W, Zhang M, Yang X. Pseudo-Kaposi’s Sarcoma: A Rare Case and Review. Clin Cosmet Investig Dermatol. 2023;16:1319–1323
.
4. Rongioletti F, Rebora A. Cutaneous reactive angiomatoses: Patterns and classification of reactive vascular proliferation. J Am Acad Dermatol. 2003;49(5):887–96.
5. Lauck K, Nguyen QB, Klimas N, Rogge M. Acroangiodermatitis presenting as unilateral hypertrophic verrucous plaques. Dermatol Online J. 2022;28.
6. Mehta A, Pereira R, Nayak C, Dhurat R. Acroangiodermatitis of mali: A rare vascular phenomenon. Indian J Dermatol Venereol Leprol. 2010;76(5).